Guide | Understanding Classic CAH: Managing the Challenges of Androgen Imbalance
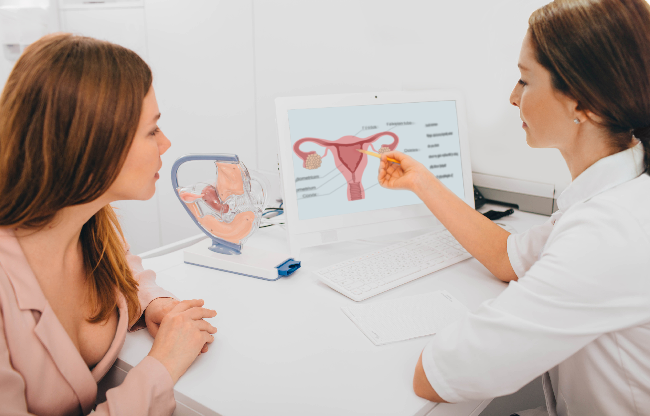
Congenital adrenal hyperplasia (CAH) is an inherited condition affecting the adrenal glands above the kidneys. These glands produce three hormones essential to normal function: cortisol, aldosterone, and androgens. Cortisol helps the body respond to stress and regulates blood pressure, energy, and blood sugar; aldosterone controls salt and water balance, blood volume, and blood pressure; and androgens, mainly testosterone, initiate and support normal growth and development during puberty.
Cause of classic CAH
Classic CAH results from a gene defect that impairs an enzyme called 21-hydroxylase, which is essential for producing cortisol and aldosterone. A person must inherit one defective gene from each parent to have CAH.
Two subtypes of classic CAH
Classic CAH has two main subtypes: salt-wasting CAH and simple virilizing CAH.
Salt-wasting CAH: The most severe type. The adrenal glands cannot produce enough aldosterone, disrupting sodium levels and potentially causing dehydration, low blood pressure, or death. Androgen levels are excessive and cortisol levels are low.
Simple virilizing CAH: This type also affects the balance of aldosterone, cortisol, and androgens, but the imbalance and symptoms are milder than in salt-wasting CAH.
Symptoms of classic CAH
High androgen levels can cause symptoms related to sexual development, including:
At birth, female infants may have virilized external genitalia, while internal reproductive organs are generally female.
Male infants may have an enlarged penis.
Early signs of puberty, including body or facial hair, voice changes, and severe acne.
Women and people assigned female at birth (AFAB) may develop masculine characteristics, such as greater muscle mass, a deeper voice, and facial hair.
Rapid growth followed by shorter adult height.
Irregular periods or infertility.
Salt-wasting CAH may cause a life-threatening adrenal crisis, with dehydration, low blood sugar, low blood sodium, or an irregular heartbeat.
Diagnosis and testing
During pregnancy, families with a child who has CAH may use amniocentesis or chorionic villus sampling (CVS) to test for the CAH gene defect.
Newborns are screened using a heel-prick blood sample. Children and adults may be diagnosed through a physical examination, blood and urine tests, X-rays, and genetic testing.
Treatment
CAH has no cure, but appropriate treatment can relieve symptoms. Care usually includes medication, hormone monitoring, and sometimes surgery.
Medication: Children generally require daily prescriptions including salt (sodium chloride) supplements, cortisol replacement such as hydrocortisone, prednisone, or dexamethasone, and a mineralocorticoid.
Surgery: Children assigned female at birth may undergo external genital surgery to reduce clitoral size and reconstruct the vaginal opening, commonly at around 6 months of age.
Other care: Options include treatment to delay puberty, medication to regulate menstruation, and nutritional and mental health support.
Emerging treatments
Researchers are studying modified-release corticosteroids, continuous corticosteroid delivery through a subcutaneous pump, and treatments targeting the hypothalamic-pituitary-adrenal (HPA) axis. These may improve hormone control.
Complications
People with salt-wasting CAH are more likely to develop electrolyte imbalance, irregular heartbeat, and cardiac arrest. Genital reconstructive surgery may also carry risks such as infection, bleeding, and scarring.
Living with CAH and support
Although CAH cannot be cured, early diagnosis and continued treatment generally support a healthy life. Lifelong medication and regular monitoring are needed to control symptoms. Mental health support is also important for managing social and emotional challenges.
Conclusion
Classic CAH is lifelong, but appropriate treatment and monitoring can support a relatively normal quality of life. Support from family and the medical team is central to managing the condition.















Guide | Understanding Classic CAH: Managing the Challenges of Androgen Imbalance
Guide | Understanding Classic CAH: Managing the Challenges of Androgen Imbalance
Congenital adrenal hyperplasia (CAH) is an inherited condition affecting the adrenal glands above the kidneys. These glands produce three hormones essential to normal function: cortisol, aldosterone, and androgens. Cortisol helps the body respond to stress and regulates blood pressure, energy, and blood sugar; aldosterone controls salt and water balance, blood volume, and blood pressure; and androgens, mainly testosterone, initiate and support normal growth and development during puberty.
Cause of classic CAH
Classic CAH results from a gene defect that impairs an enzyme called 21-hydroxylase, which is essential for producing cortisol and aldosterone. A person must inherit one defective gene from each parent to have CAH.
Two subtypes of classic CAH
Classic CAH has two main subtypes: salt-wasting CAH and simple virilizing CAH.
Salt-wasting CAH: The most severe type. The adrenal glands cannot produce enough aldosterone, disrupting sodium levels and potentially causing dehydration, low blood pressure, or death. Androgen levels are excessive and cortisol levels are low.
Simple virilizing CAH: This type also affects the balance of aldosterone, cortisol, and androgens, but the imbalance and symptoms are milder than in salt-wasting CAH.
Symptoms of classic CAH
High androgen levels can cause symptoms related to sexual development, including:
At birth, female infants may have virilized external genitalia, while internal reproductive organs are generally female.
Male infants may have an enlarged penis.
Early signs of puberty, including body or facial hair, voice changes, and severe acne.
Women and people assigned female at birth (AFAB) may develop masculine characteristics, such as greater muscle mass, a deeper voice, and facial hair.
Rapid growth followed by shorter adult height.
Irregular periods or infertility.
Salt-wasting CAH may cause a life-threatening adrenal crisis, with dehydration, low blood sugar, low blood sodium, or an irregular heartbeat.
Diagnosis and testing
During pregnancy, families with a child who has CAH may use amniocentesis or chorionic villus sampling (CVS) to test for the CAH gene defect.
Newborns are screened using a heel-prick blood sample. Children and adults may be diagnosed through a physical examination, blood and urine tests, X-rays, and genetic testing.
Treatment
CAH has no cure, but appropriate treatment can relieve symptoms. Care usually includes medication, hormone monitoring, and sometimes surgery.
Medication: Children generally require daily prescriptions including salt (sodium chloride) supplements, cortisol replacement such as hydrocortisone, prednisone, or dexamethasone, and a mineralocorticoid.
Surgery: Children assigned female at birth may undergo external genital surgery to reduce clitoral size and reconstruct the vaginal opening, commonly at around 6 months of age.
Other care: Options include treatment to delay puberty, medication to regulate menstruation, and nutritional and mental health support.
Emerging treatments
Researchers are studying modified-release corticosteroids, continuous corticosteroid delivery through a subcutaneous pump, and treatments targeting the hypothalamic-pituitary-adrenal (HPA) axis. These may improve hormone control.
Complications
People with salt-wasting CAH are more likely to develop electrolyte imbalance, irregular heartbeat, and cardiac arrest. Genital reconstructive surgery may also carry risks such as infection, bleeding, and scarring.
Living with CAH and support
Although CAH cannot be cured, early diagnosis and continued treatment generally support a healthy life. Lifelong medication and regular monitoring are needed to control symptoms. Mental health support is also important for managing social and emotional challenges.
Conclusion
Classic CAH is lifelong, but appropriate treatment and monitoring can support a relatively normal quality of life. Support from family and the medical team is central to managing the condition.
Source:
Collected online